

Introduction
molUP is a VMD extension that provides a simple manner for loading and saving Gaussian files, and analyze related results. molUP provides a graphical interface for VMD where the users can load and save chemical structures in the Gaussian file formats. This extension includes a set of tools to set up any calculation supported by Gaussian, including ONIOM; analyze energies through interactive plots; animate vibrational frequencies; draw the vectors associated with those frequencies; modify bonds, angles, and dihedrals; and collect bibliographic information on the employed methods.
Features
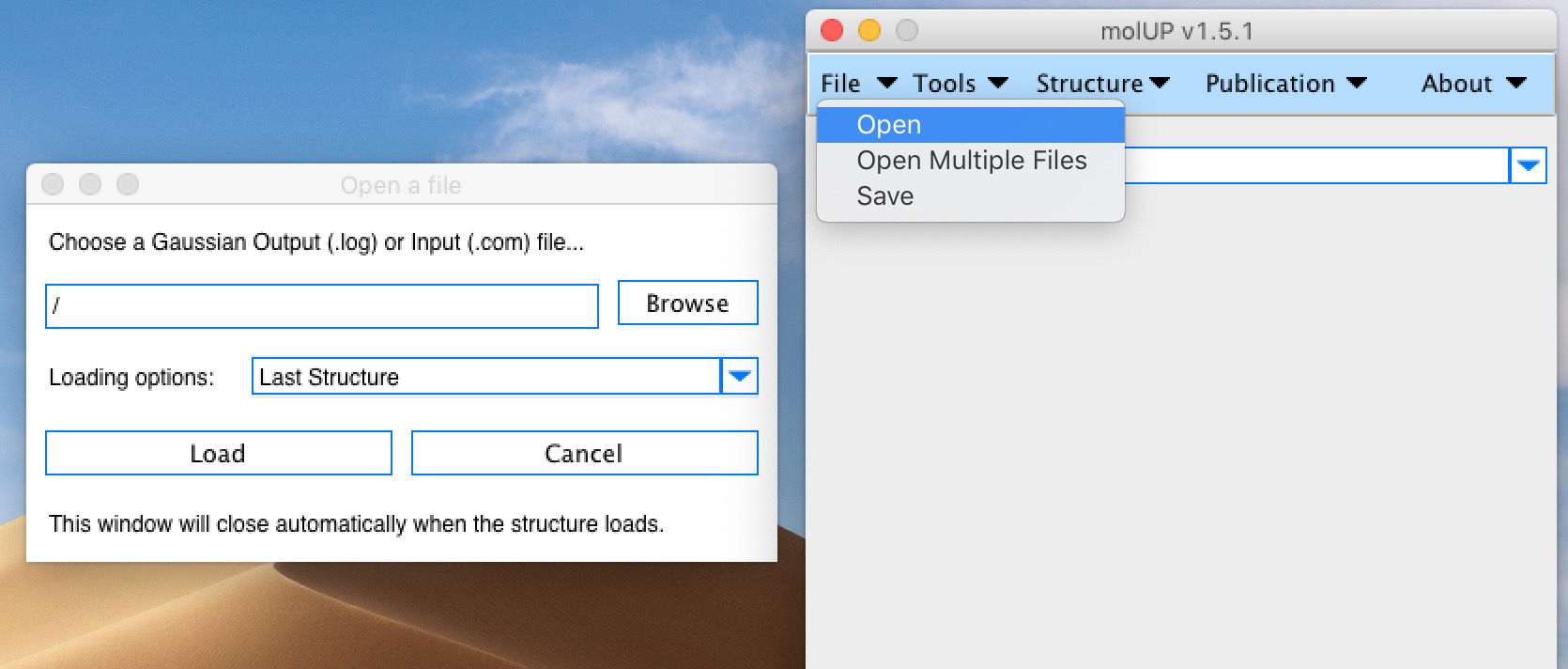
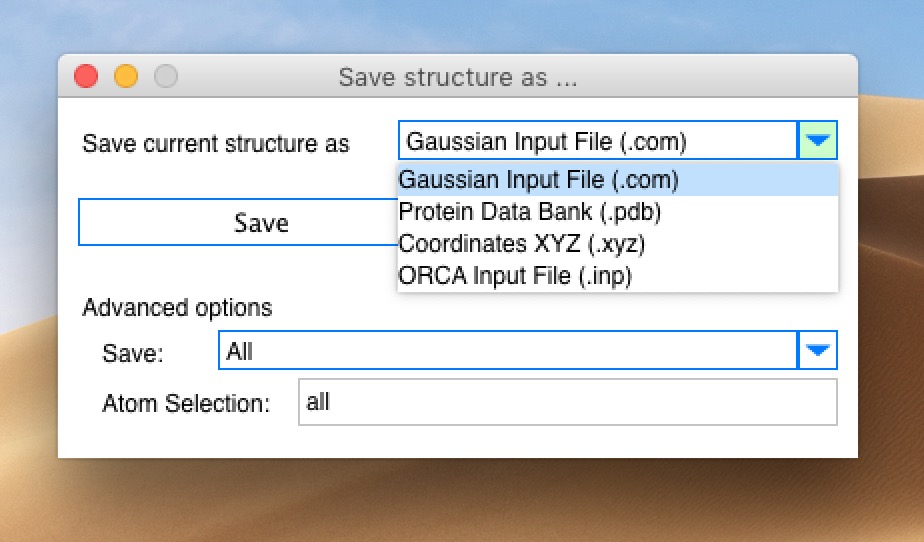
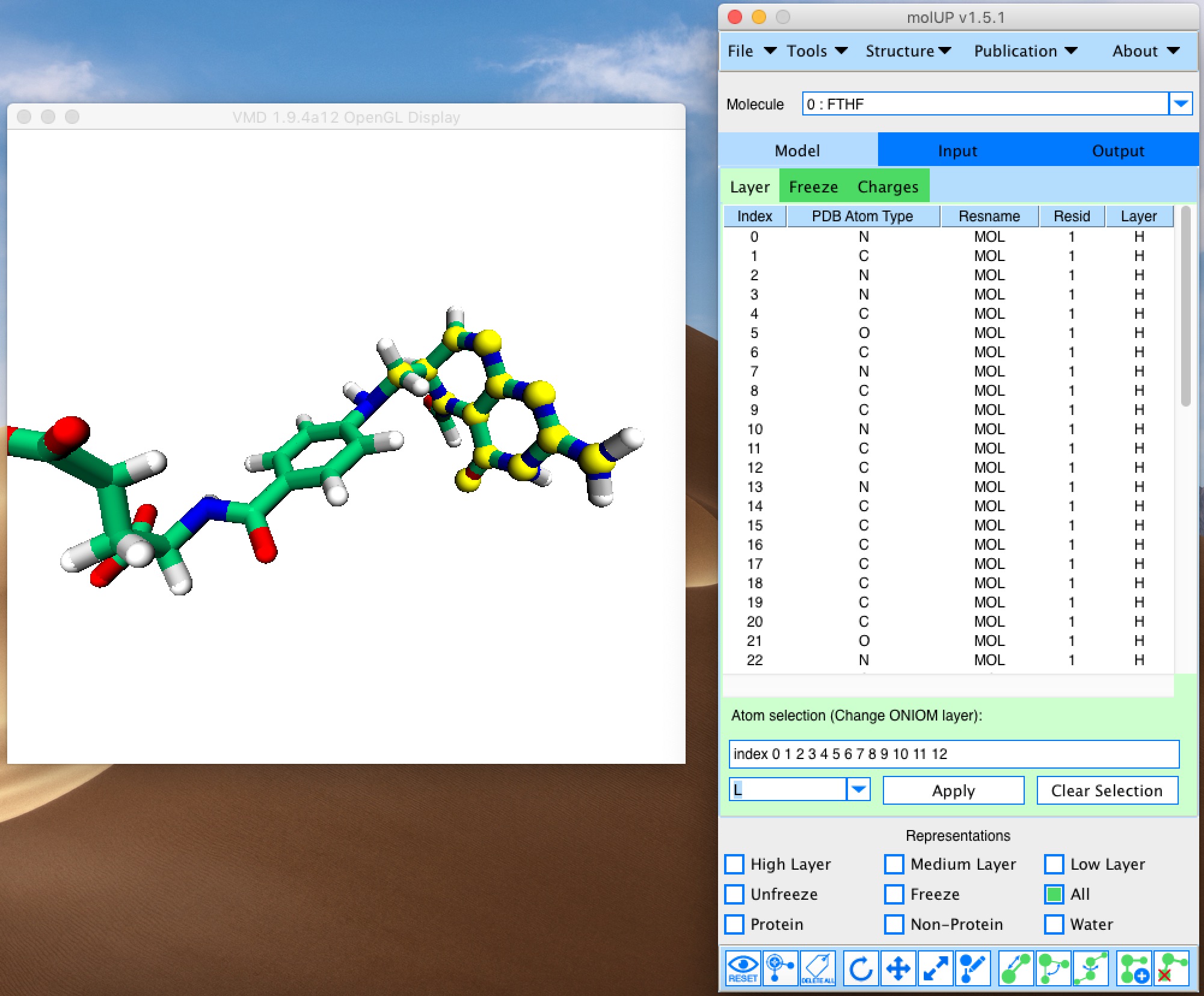
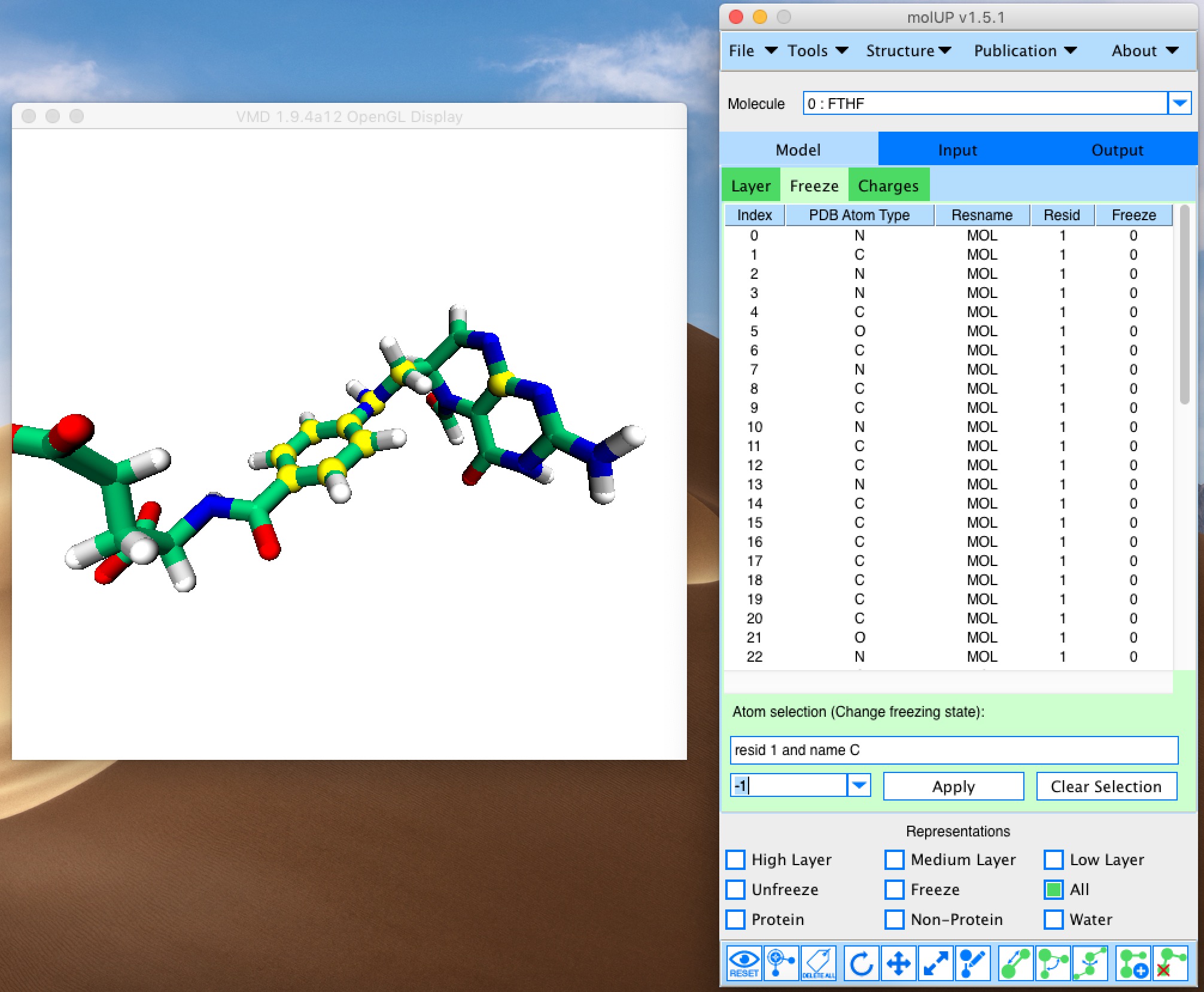
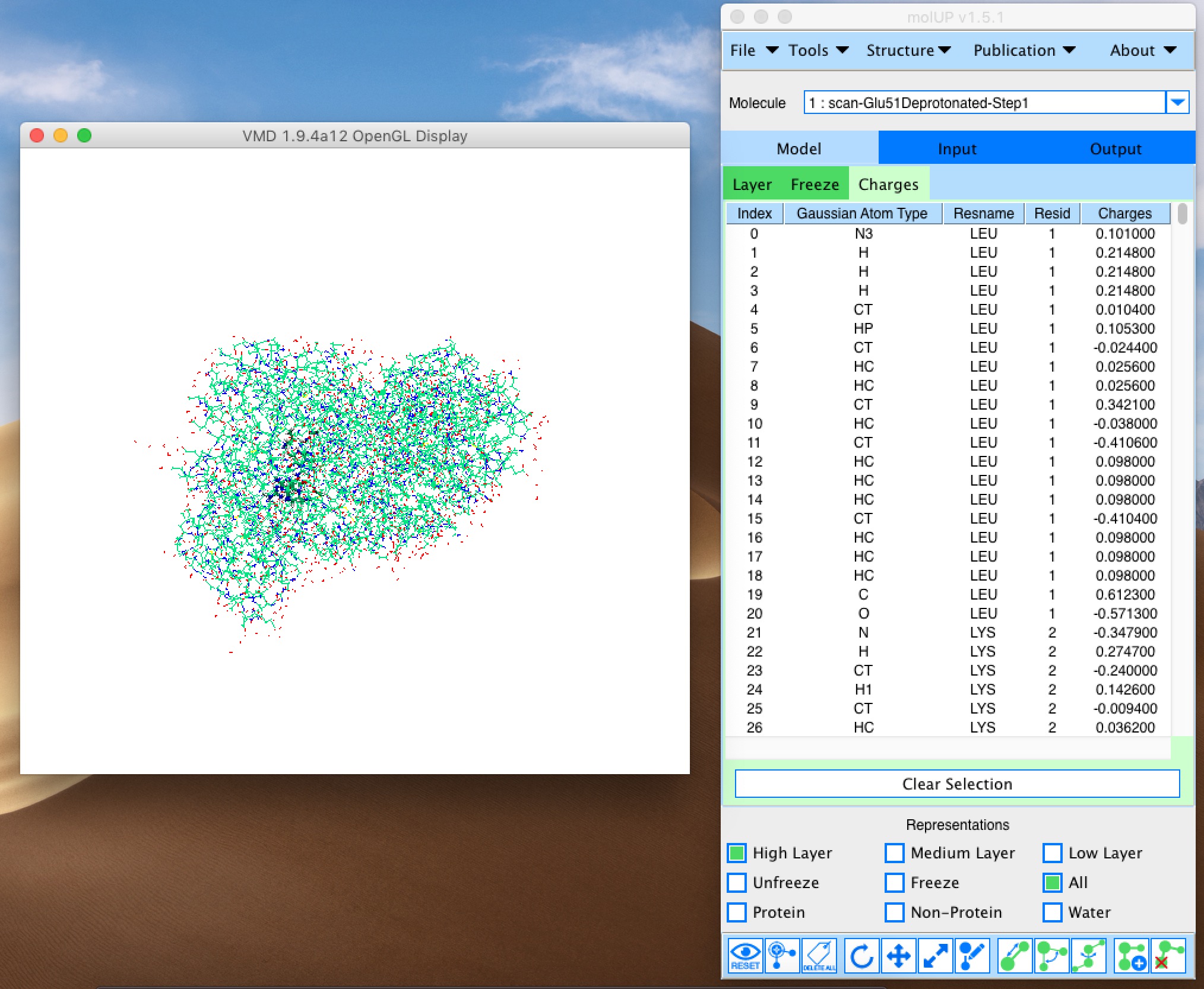
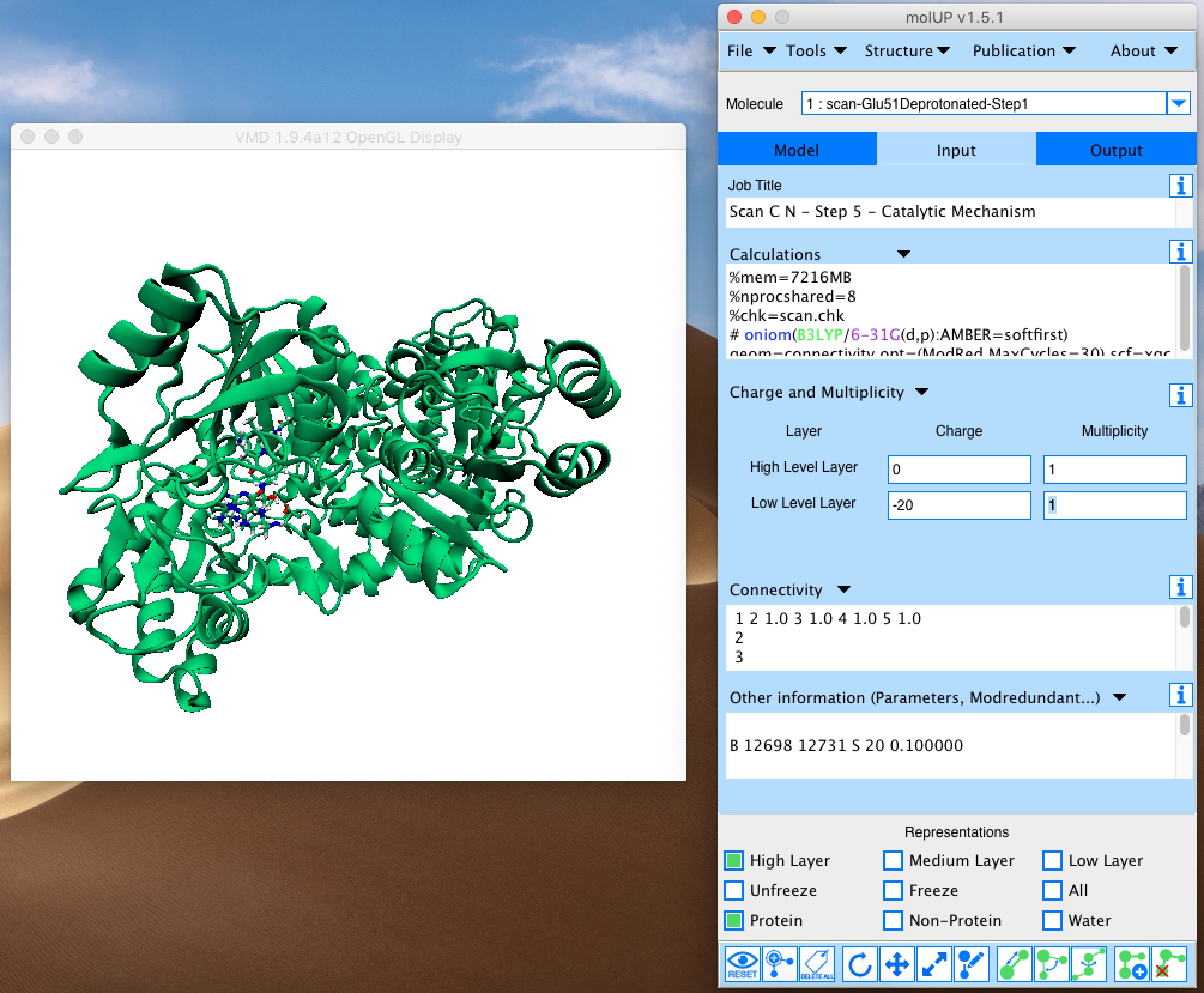
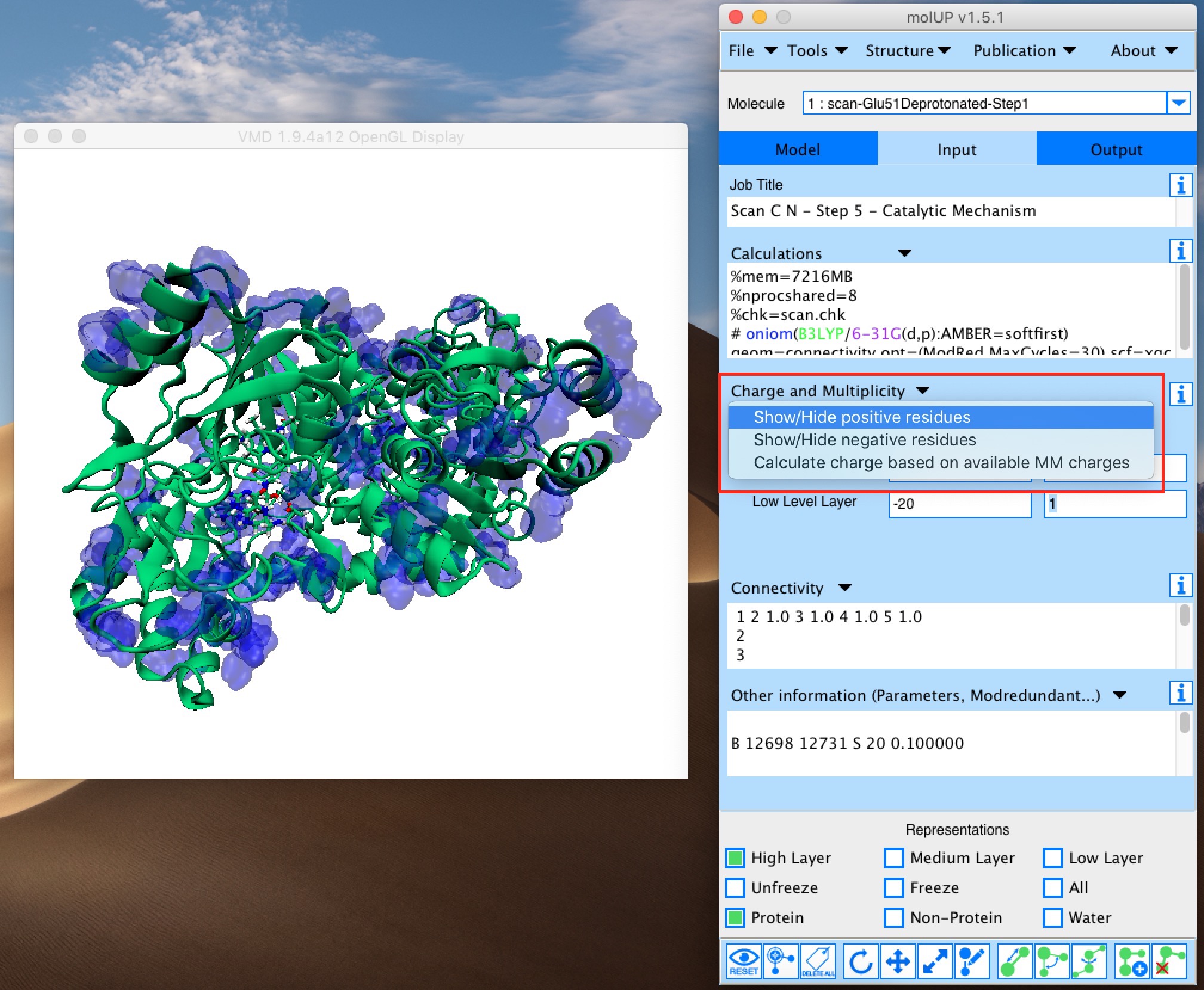
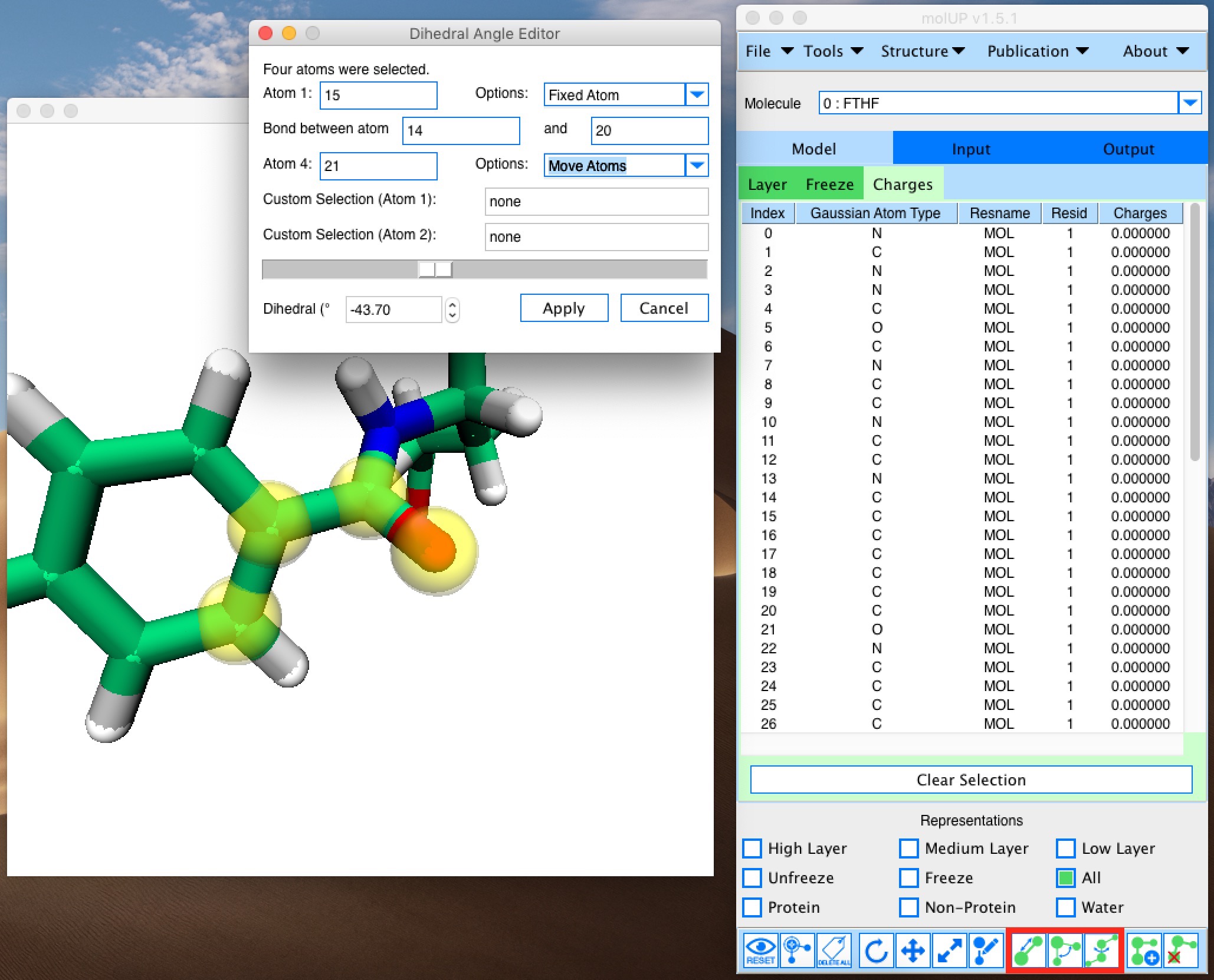
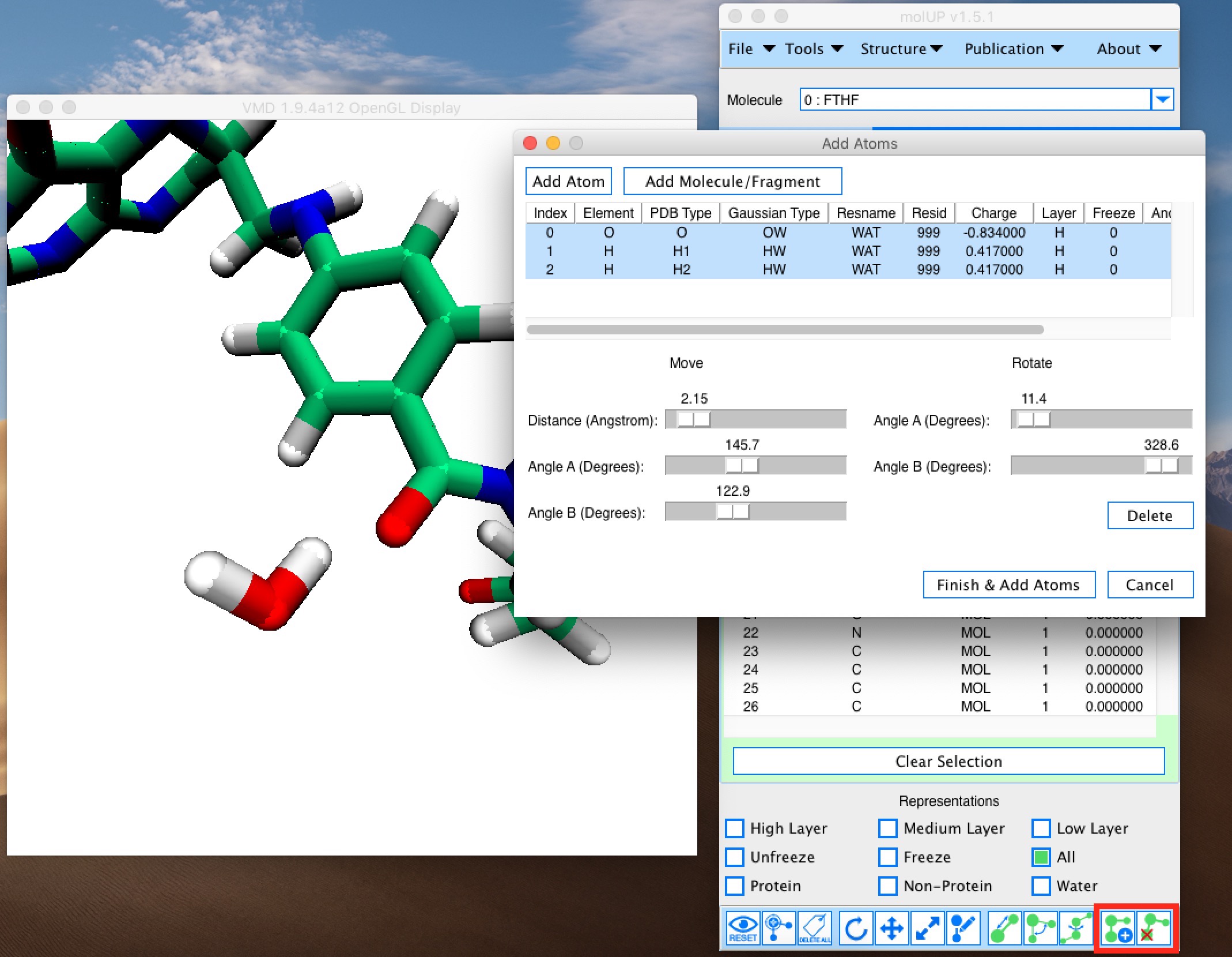
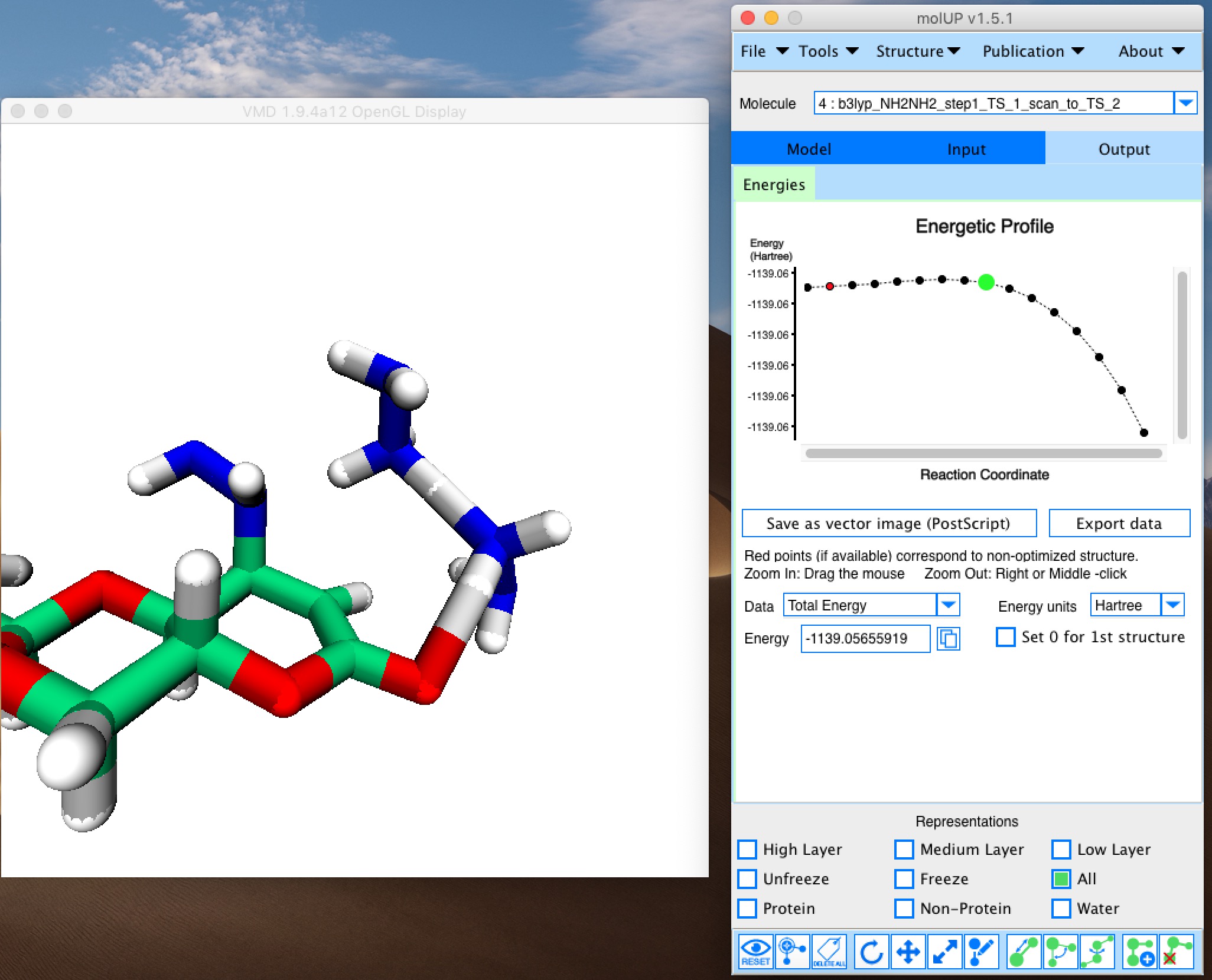
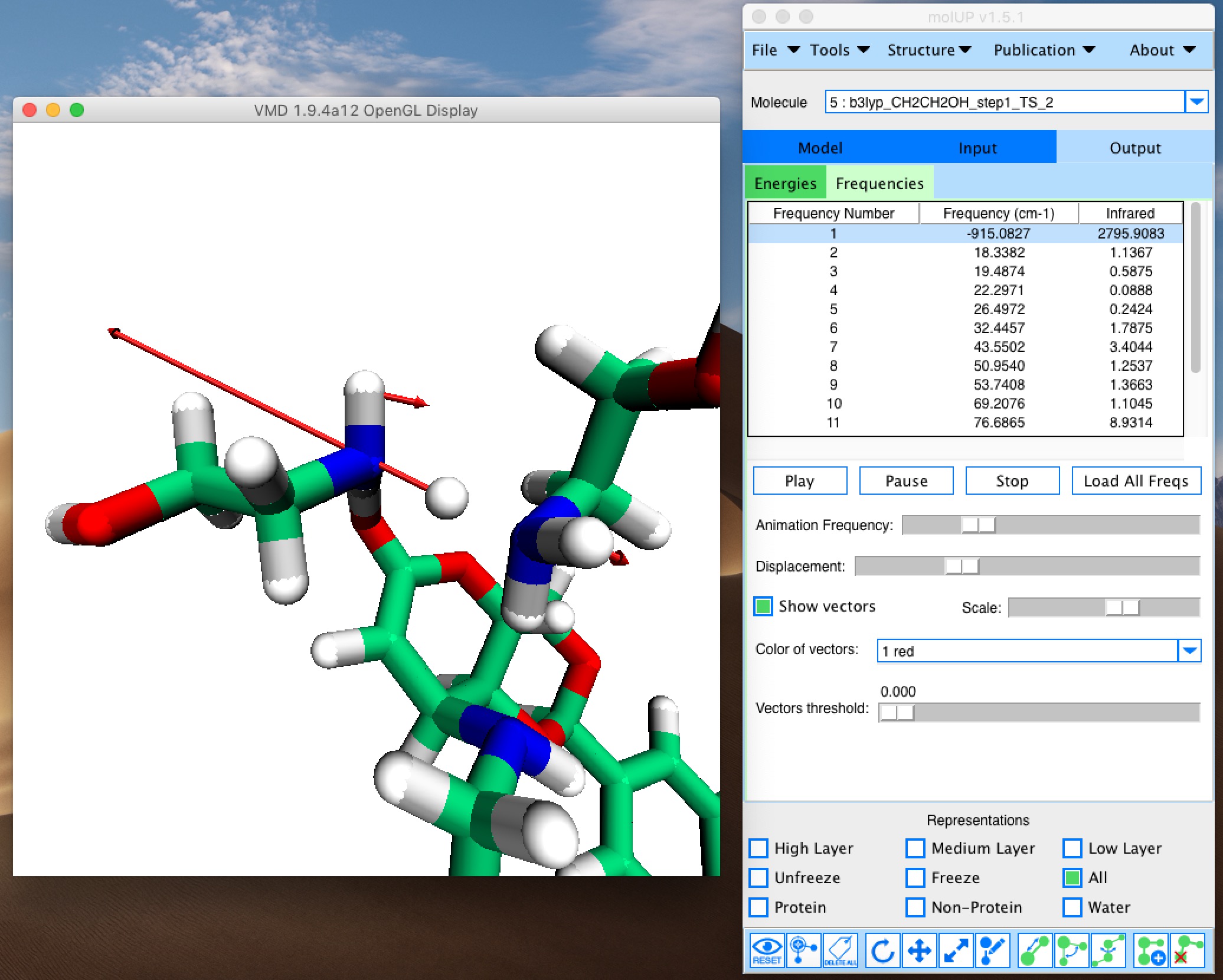
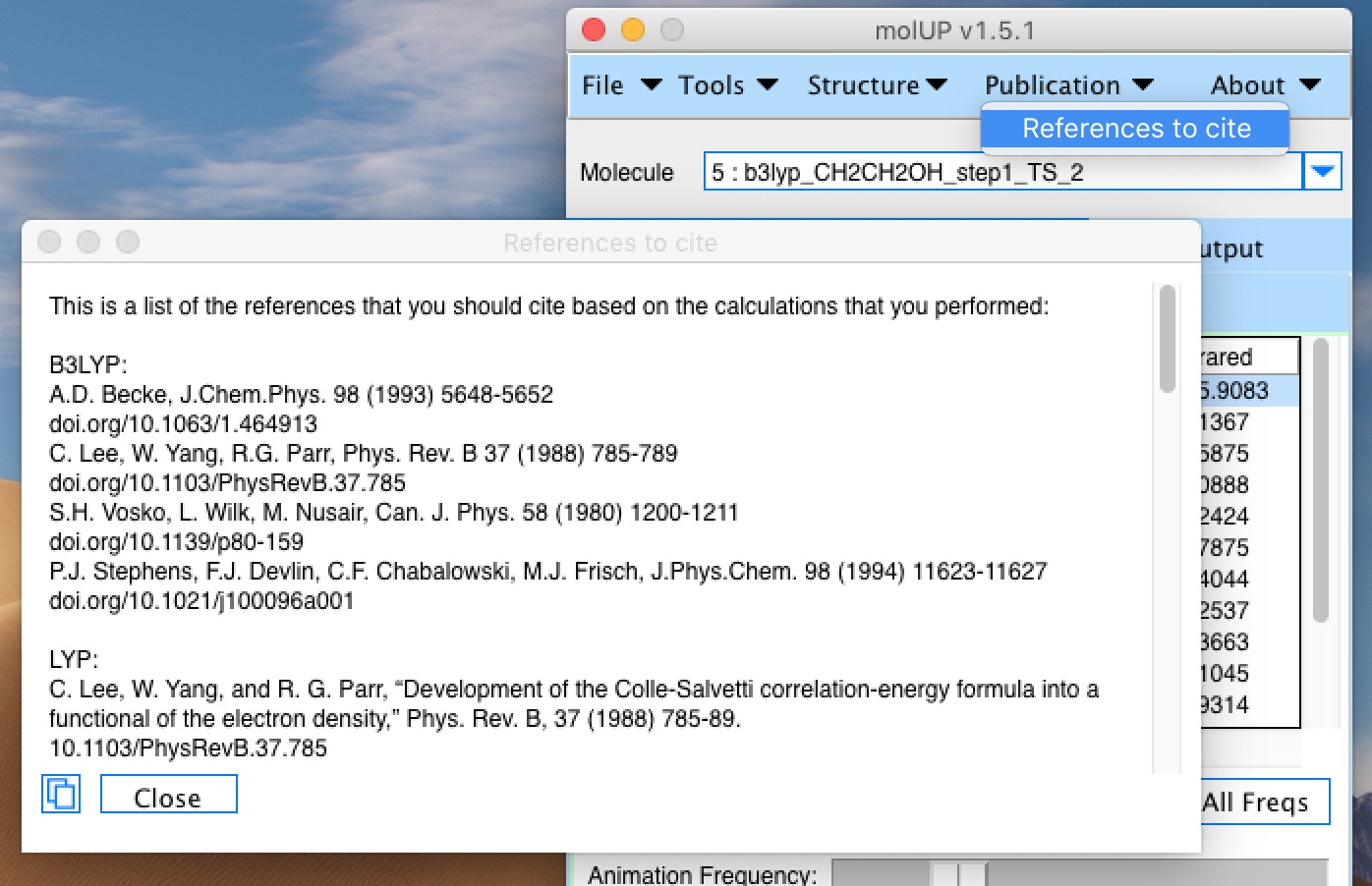
Tutorials
How to start a QM/MM study from a Molecular Dynamics (MD) simulation? [See Tutorial]
Detailed tutorials:
molUP Tutorial 1 – Start a QM/MM study
molUP Tutorial 2 – Load a Gaussian output and prepare a scan
Minimum Requirements
Operating System: macOS, Linux, or Windows.
Visual Molecular Dynamics (VMD) 1.9.3 (Free Download) or later.
Installation
Automatic Installation
You could easily install molUP through the VMD Store. 
Manual Installation
Alternatively, you could install molUP manually:
- Download or Clone the repository.
- Copy the molUP directory to a location on your computer. (Get that location!)
- Copy the text inside “install.txt” file and paste on your .vmdrc file (macOS and Linux ) or vmd.rc file (Windows).
- Replace the string “$::vmdStorePath/plugins/molUP” by your installation location (Step 2).
- Save the .vmdrc or vmd.rc file.
- Restart VMD.
(Optional) Run molUP from bash
molUP supports now the loading of Gaussian files directly from your command line (available for macOS and Linux).
Tutorial:
- Add the following line to your .bashrc (Linux) or .bash_profile (macOS) file. (This file is located in the HOME directory) You have to edit the “path of the bashScript.tcl file” field by the complete path of this file that is located in the molUP installation directory: “molUP/lib/bashScript.tcl”
alias molUP='vmd -e "path of the bashScript.tcl file" -args'
- Call molUP from the command line:
Open a Gaussian file using the standard procedure:
molUP "Gaussian file"
Open a Gaussian output file reading all available structures:
molUP "Gaussian file" -all
Report Bugs/Suggestions
If you experienced any type of issue, please contact us to fix it as soon as possible. (Provide the error message and the file that causes it.)
If you have any idea of new features, please let us know to try the implementation in a future version.
E-Mail: [email protected]
Troubleshooting
macOS sed and gsed
On macOS, the sed command needs to point to GNU-sed (gsed). Using brew, you can easily install GNU-sed a native sed on macOS:
brew uninstall gnu-sed && brew install gnu-sed --with-default-names
Examples of errors that occur in this situation:
------------------------------------------------------
sed: 1: "5 {p; :loop n; q; b loop}": unexpected EOF (pending }'s)
sed: 1: "5 {p; :loop n; q; b loop}": unexpected EOF (pending }'s)
while executing
"exec $molUP::sed -n "$lineNumberTitle {p; :loop n; q; b loop}" $molUP::path"
(procedure "molUP::loadGaussianInputFile" line 5)
invoked from within
"molUP::loadGaussianInputFile"
(procedure "molUP::loadButton" line 9)
invoked from within
"molUP::loadButton $molUP::fileExtension"
invoked from within
".molUP.openFile.frame.back.buttonLoad invoke "
invoked from within
".molUP.openFile.frame.back.buttonLoad instate {pressed !disabled} { .molUP.openFile.frame.back.buttonLoad state !pressed; .molUP.openFile.frame.back.b..."
(command bound to event)
------------------------------------------------------
list element in quotes followed by ":" instead of space
list element in quotes followed by ":" instead of space
while executing
"llength [lindex $molUP::structureGaussian 0]"
(procedure "molUP::loadGaussianOutputFile" line 79)
invoked from within
"molUP::loadGaussianOutputFile lastStructure"
(procedure "molUP::loadButton" line 19)
invoked from within
"molUP::loadButton $molUP::fileExtension"
invoked from within
".molUP.openFile.frame.back.buttonLoad invoke "
invoked from within
".molUP.openFile.frame.back.buttonLoad instate {pressed !disabled} { .molUP.openFile.frame.back.buttonLoad state !pressed; .molUP.openFile.frame.back.b..."
(command bound to event)
Internet connection causing VMD Store installation issues
If you cannot install molUP through VMD Store, please check if you are able to access github.com. Some countries/networks could block access to specific domains as GitHub, precluding the installation process.
Paths on Windows OS
If you experienced some kind of error loading files, please check the vmdStore.rc or vmd.rc files. Sometimes, Windows OS provides different types of paths for the some directory, causing issues. For example, the following line:
variable vmdStorePath "/users/name/vmdStore"
should be changed to:
variable vmdStorePath "C:/Users/name/vmdStore"
Examples of errors that occur in this situation:
unknown color name "/users/eddie/vmdStore/plugins/molUP/user/references.txt:"
unknown color name "/users/eddie/vmdStore/plugins/molUP/user/references.txt:"
(processing "-foreground" option)
invoked from within
"$pathName tag configure "[subst $color]" -foreground "$color""
(procedure "molUP::checkTags" line 8)
invoked from within
"molUP::checkTags .molUP.frame0.major.mol$mol.tabs.tabInput.keywordsText"
(procedure "molUP::updateStructures" line 27)
invoked from within
"molUP::updateStructures"
(procedure "molUP::loadButton" line 21)
invoked from within
"molUP::loadButton $molUP::fileExtension"
invoked from within
".molUP.openFile.frame.back.buttonLoad invoke "
invoked from within
".molUP.openFile.frame.back.buttonLoad instate {pressed !disabled} { .molUP.openFile.frame.back.buttonLoad state !pressed; .molUP.openFile.frame.back.b..."
(command bound to event)
If you used molUP, please cite
H. S. Fernandes, M. J. Ramos, N. M. F. S. A. Cerqueira J. Comput. Chem. 2018, 39, 1344–1353. DOI: 10.1002/jcc.25189
Changelog
See here.