
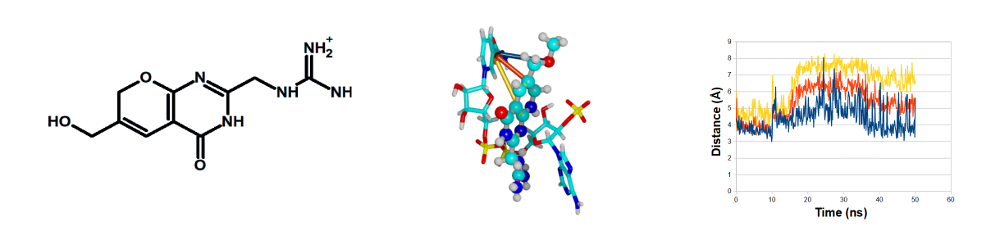
We are glad to announce that our research on the development of novel inhibitors of R67 dihydrofolate reductase (DHFR) has just been published in Antibiotics (IF: 4.639). In this study, the acid-base and redox characteristics of several newly-designed structural analogues of the enzyme-substrate were first studied using quantum-mechanical methods. The few analogues proven by these methods to be stable against modification by the target enzyme were then docked into it, and the stability of their binding modes were analysed through triplicate molecular dynamics simulations. One of them (([6-(methoxymethyl)-4-oxo-3,7-dihydro-4H-pyrano[2,3-d]pyrimidin-2-yl]methyl-guanidinium)) afforded very stable binding against both the atypical R67 DHFR
and the canonical prokaryotic and eukaryotic DHFR. This research therefore shows that this novel molecule is a very promising candidate towards eventual clinical applications both against infectious bacteria or against fast-dividing, tumoral, eukaryotic cells. Additionally, our simulations afford extra insight regarding the factors underlying the interaction between R67 DHFR and its natural substrate, and its relatively low catalytic efficiency.
Computational Development of Inhibitors of Plasmid-Borne Bacterial Dihydrofolate Reductase
Pedro J. Silva
Antibiotics 2022, 11(6), 779 | DOI: 10.3390/antibiotics11060779
Resistance to trimethoprim and other antibiotics targeting dihydrofolate reductase may arise in bacteria harboring an atypical, plasmid-encoded, homotetrameric dihydrofolate reductase, called R67 DHFR. Although developing inhibitors to this enzyme may be expected to be promising drugs to fight trimethoprim-resistant strains, there is a paucity of reports describing the development of such molecules. In this manuscript, we describe the design of promising lead compounds to target R67 DHFR. Density-functional calculations were first used to identify the modifications of the pterin core that yielded derivatives likely to bind the enzyme and not susceptible to being acted upon by it. These unreactive molecules were then docked to the active site, and the stability of the docking poses of the best candidates was analyzed through triplicate molecular dynamics simulations, and compared to the binding stability of the enzyme–substrate complex. Molecule 32 ([6-(methoxymethyl)-4-oxo-3,7-dihydro-4H-pyrano[2,3-d]pyrimidin-2-yl]methyl-guanidinium) was shown by this methodology to afford extremely stable binding towards R67 DHFR and to prevent simultaneous binding to the substrate. Additional docking and molecular dynamics simulations further showed that this candidate also binds strongly to the canonical prokaryotic dihydrofolate reductase and to human DHFR, and is therefore likely to be useful to the development of chemotherapeutic agents and of dual-acting antibiotics that target the two types of bacterial dihydrofolate reductase.